1.1 Introduction
Breast cancer (BC) is the most frequently diagnosed cancer in women worldwide, with 1.7 million new cases diagnosed in 2012 []. Luminal A BCs are ER+ and/or PR+ but HER2. These are the most commonly seen BCs and have the best prognosis. Luminal B BCs are ER+ and/or PR+ and sometimes HER2+. These tumors have a higher proliferative index and are more aggressive. HER2+ BCs are ER and PR but HER2+. This subtype usually presents at a younger age with a poorer tumor grade and lymph node involvement, but the prognosis has improved dramatically since the clinical implementation of Herceptin, an anti-HER2 antibody. About 20% of BCs are triple negative (TNBC) or basal-like, that is, they are ER, PR, and HER2. These tumors are often aggressive, have poorer prognosis, and lack any targeted therapies.
Research has focused extensively on the role of cell surface receptors like HER2 in the pathobiology of BC. There are numerous families of cell surface receptors, like receptor tyrosine kinases (RTKs), one example being HER2, and G protein-coupled receptors, that sense extracellular cues and transmit them into intracellular messages that regulate cell growth, proliferation, survival, migration, and differentiation. These receptors are often deregulated in BC and lead to tumor growth and metastasis. This chapter will focus on the identification of the receptors most often deregulated in BC, the common signaling pathways they activate, and the crosstalk that links them to one another.
1.2 RTKs and Their Downstream Signaling Targets
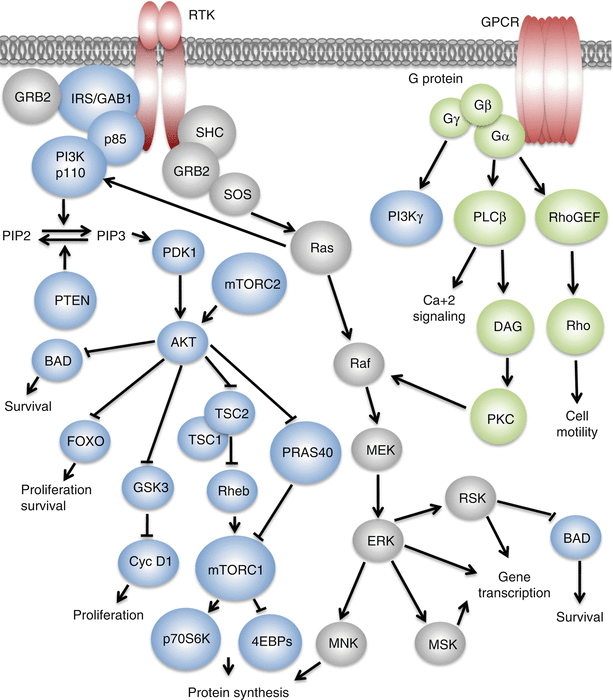
RTKs are cell surface receptors found on a diversity of cell types. All RTKs comprise an N-terminal extracellular ligand-binding domain, a single-pass transmembrane domain, and a C-terminal tyrosine kinase domain. Ligand binding induces a conformational change leading to the receptor homo- or heterodimerization and the consequent autophosphorylation of a series of tyrosine residues in the C-terminal tail. The phosphorylated tyrosines then act as docking sites for the SRC homology 2 (SH2) and phosphotyrosine-binding (PTB) domain-containing proteins, many of which are shared by the different RTKs. The RTK signaling program converges on the two major signaling pathways, namely, the phosphoinositide 3-kinase-protein kinase B/AKT (PI3K-PKB/AKT) and the rat sarcoma-mitogen-activated protein kinase/ERK (Ras-MAPK/ERK), that go on to regulate critical cellular processes like cell growth, proliferation, differentiation, migration, and apoptosis (Fig. ).
Fig. 1.1
RTK and GPCR signaling networks
One of the SH2 domain-containing effectors of RTKs is the regulatory subunit of the class I PI3K (p85), which when bound to the activated RTK or one of its tyrosine phosphorylated adaptors relieves its inhibition of the p110 catalytic subunit of PI3K, thereby leading to its activation [].
The Ras-ERK pathway is the other major signaling network that is modulated by the RTKs. The Src homology 2 domain-containing (SHC) and the growth factor receptor bound 2 (GRB2) are the main adaptor proteins that link the activated RTKs to the Ras-ERK pathway []. The Ras-ERK pathway thus controls diverse cellular processes including cell growth, proliferation, differentiation, migration, and apoptosis.
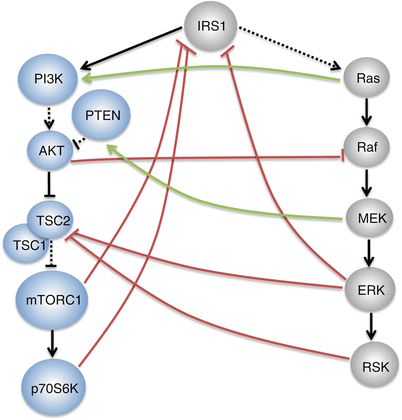
In addition to the parallel activation immediately downstream of the receptors, coordination between the PI3K-AKT and the Ras-ERK signaling pathways can also be achieved by the interaction of activated Ras with the PI3K p110 catalytic subunit, independently of p85, leading to the PI3K-AKT pathway activation (Fig. ]. Thus, multiple feedback loops and crosstalk between the PI3K-AKT and the Ras-ERK pathways orchestrate the dynamic and intricate, context-dependent effects of multiple growth factors through their cognate RTKs.
Fig. 1.2
Crosstalk between the PI3K-AKT and the Ras-ERK signaling pathways. Green lines indicate activation and red lines indicate inhibition. Solid black lines indicate a direct interaction, while dashed black lines indicate an indirect interaction
1.3 RTKs Often Deregulated in BC
The deregulation of RTK signaling plays an important role in the pathophysiology of many cancers, including BC []. Several mechanisms lead to the deregulation of RTK signaling, including RTK gene amplifications, activating mutations, protein overexpression, ligand overexpression or hyperactivation, and crosstalk with other cellular signaling components. Members of the ERBB family, MET, the insulin receptor (INSR), and the insulin-like growth factor receptor (IGF1R) are RTKs that are most often deregulated in BC.
1.3.1 HER2
The amplification of the HER2 gene, a member of the ERBB family of RTKs, is seen in approximately 20% of BCs, and HER2 overexpression correlates with a worse BC prognosis [].
Like other prototypical RTKs, all ERBBs can form functional homo- or heterodimers, with the exception of HER2, which does not appear to bind a ligand, and ERBB3, which is impaired in the intrinsic kinase activity and thus cannot form functional homodimers []. Alternatively, ERBBs can activate the PI3K pathway through Ras. Together with the multitude of ligands, the different combinations of receptor dimers, and the unique C-terminal tails, this family of RTKs is capable of regulating diverse cellular processes implicated in cell growth, proliferation, differentiation, migration, and apoptosis.
1.3.2 MET
The hepatocyte growth factor receptor or MET is another RTK that is overexpressed in about 20% of BCs, particularly in the basal-like TNBCs [].
1.3.3 INSR
The INSR is overexpressed in as many as 80% of BCs and is associated with poor survival [].
On the cell surface, the INSR exists as a heterotetrameric protein comprised of two extracellular alpha subunits and two transmembrane beta subunits. The beta subunit of the receptor possesses tyrosine kinase activity, which is stimulated upon binding of the ligand to the alpha subunit [].
1.3.4 IGF1R
Close to 50% of human breast tumors express the activated form of IGF1R, and gene expression signatures consistent with IGF1R activation are associated with poor outcome in BC patients []. As a result, IGF2R may exhibit tumor suppressor properties by decreasing the bioactivity of IGFII and indirectly modulating signaling by IGF1R.
Due to their homology and strong structural similarities, the INSR and IGF1R have the ability to form hybrid receptors composed of one hemireceptor of each type. In addition, the two INSR isoforms can also combine to form hybrids, generating the potential for multiple insulin and IGF-sensitive receptors (INSR-A, INSR-B, INSR-A/B, IGF1R, INSR/IGF1R) to be expressed by a single cell. Hybrid receptors appear to bind IGFI with a higher affinity than insulin, and they exhibit different ligand specificities depending on the INSR isoform present. For example, INSR-A/IGF1R hybrids bind IGFI, IGFII, and insulin, while INSR-B/IGF1R hybrids typically bind IGF1 []. Consequently, human BC cells are highly sensitive to the growth-promoting effects of insulin and IGFs, and INSR/IGF1R expression may be a key event in tumor development and growth.