1.1 Introduction
Scleroderma, or systemic sclerosis (SSc), is a generalized connective tissue disease that is characterized by sclerotic changes in the skin and sometimes various other organ systems (Fig. ].
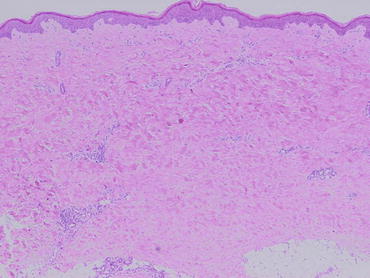
Fig. 1.1
Histopathology of the skin in a patient with SSc, showing thickened collagen bundles
Fibrosis is the most characteristic pathological hallmark of SSc. Fibrosis is a complex biological process involving an acute inflammatory response. Transient activation of fibroblasts to proliferate and produce elevated quantities of ECM is essential to fibrosis. It is likely that such transient fibroblast activation is regulated by a variety of cytokines produced by infiltrating platelets, monocytes/macrophages, T lymphocytes, and other inflammation-associated cells [].
Increasing evidence suggests that activation of lesional fibroblasts contributes to the fibrotic process [].
The mechanism of fibroblast activation in SSc is presently unknown. However, many of the characteristics of SSc fibroblasts resemble those of healthy fibroblasts stimulated by TGF- [], suggesting that TGF- is a key mediator of tissue fibrosis in SSc.
The most potent profibrotic stimulus to fibroblasts is TGF-. The TGF- superfamily, which includes the prototypic factor TGF-1, has a shared structure, similar signaling pathways, and an overlap of biological effects. TGF- is a 25 kD homodimeric polypeptide, which participates in a broad array of biological activities such as normal development, wound repair, and pathological processes []. TGF- regulates multiple cellular functions including inhibition and stimulation of cell growth, cell death or apoptosis, and cellular differentiation.
1.2 TGF-
1.2.1 TGF- Superfamily, Structure, and Activation
The TGF- superfamily includes the various forms of TGF-, bone morphogenic protein (BMP), activin, nodals, the anti-Mullerian hormone, and many other structurally related factors [].
Although in vitro effects of TGF- isoforms seem to be similar, mice lacking TGF- isoforms revealed that each TGF- isoform plays an independent and non-redundant role in vivo. Disruption of the TGF-1 gene results in a wasting syndrome accompanied by a multifocal, mixed inflammatory cell response and tissue necrosis, leading to organ failure and death, which indicates a critical role of TGF-1 in immune regulation [].
Enhanced expression of TGF- has been well demonstrated in the tissue of SSc as well as that of a mouse model of SSc [].
1.2.2 TGF- Receptors
TGF- exerts its multiple biological actions by an interaction with two transmembrane serine/threonine kinase receptors, types I and II, that are coexpressed by most cells including mesenchymal and endothelial cells. From the structural point of view, type I and type II TGF- receptors are very similar glycoproteins, characterized by a cysteine-rich extracellular domain, a single hydrophobic transmembrane domain, and a C-terminal cytoplasmic serine/threonine kinase domain. Initiation of signaling requires the binding of TGF- to TGF- receptor type II, a constitutively active serine/threonine kinase, which results in the recruitment and phosphorylation of the type I TGF- receptor to produce a heteromeric complex that activates downstream signaling pathways []. Betaglycan, a transmembrane proteoglycan also known as TGF- receptor type III, allows high-affinity binding of TGF- to TGF- receptor type II, but does not itself transduce signal.
Increased expression of TGF- receptors has been well demonstrated in fibrosis [].
1.3 SMAD
1.3.1 SMADS Proteins
Following ligand activation, signaling from TGF- receptor type I to the nucleus occurs predominantly by phosphorylation of cytoplasmic proteins belonging to the Smad family []. Recent studies have focused on the role of Smad in tissue fibrosis.
Despite extensive research to elucidate the role of Smad proteins downstream of TGF-, few direct Smad target ECM genes have been characterized. For example, the transcriptional activation of the fibronectin gene by TGF- was shown to be regulated by a JNK-specific, Smad-independent mechanism [].
Smad proteins have been reported to be involved in the transcriptional regulation of the gene for type I collagen, which is excessively deposited in fibrosis. The human 2(I) collagen gene is upregulated at transcriptional level in fibrotic lesion [].
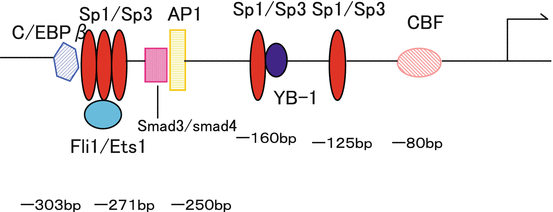
Fig. 1.2
Transcriptional regulation of the human 2(I) collagen gene. The human 2(I) collagen promoter is regulated by transcription factors Sp1/Sp3, Ets1/Fli1, C/EBP-, AP-1, Smad3/Smad4, YB-1, and CCAAT-binding factor (CBF)
Several studies showed that mice lacking Smad3 have a significantly reduced cutaneous as well as pulmonary fibrotic response []. These not only suggest the potential role of Smad7 in fibrosis, but also may lead to the development of novel approaches for treating fibrosis.
The expression levels of Smad proteins were also investigated in SSc fibroblasts [].
1.3.2 Coactivators and Corepressors of SMAD-Dependent Gene Transcription
In the nucleus, activated Smad complexes regulate various gene expressions with the recruitment of coactivators or corepressors into transcriptional complexes. The role of CREB binding protein (CBP) and p300 as essential coactivators for Smad-driven gene expression has been well demonstrated []. These impaired inhibitory effects of c-Ski/SnoN are characteristic for SSc fibroblasts and may be involved in the pathogenesis of fibrosis in SSc. However, the role of other corepressors in tissue fibrosis, especially in SSc, is still poorly understood.