Chauve Cedric - Models and Algorithms for Genome Evolution
Here you can read online Chauve Cedric - Models and Algorithms for Genome Evolution full text of the book (entire story) in english for free. Download pdf and epub, get meaning, cover and reviews about this ebook. City: London, year: 2013, publisher: Springer London : Imprint : Springer, genre: Children. Description of the work, (preface) as well as reviews are available. Best literature library LitArk.com created for fans of good reading and offers a wide selection of genres:
Romance novel
Science fiction
Adventure
Detective
Science
History
Home and family
Prose
Art
Politics
Computer
Non-fiction
Religion
Business
Children
Humor
Choose a favorite category and find really read worthwhile books. Enjoy immersion in the world of imagination, feel the emotions of the characters or learn something new for yourself, make an fascinating discovery.

- Book:Models and Algorithms for Genome Evolution
- Author:
- Publisher:Springer London : Imprint : Springer
- Genre:
- Year:2013
- City:London
- Rating:4 / 5
- Favourites:Add to favourites
- Your mark:
Models and Algorithms for Genome Evolution: summary, description and annotation
We offer to read an annotation, description, summary or preface (depends on what the author of the book "Models and Algorithms for Genome Evolution" wrote himself). If you haven't found the necessary information about the book — write in the comments, we will try to find it.
Chauve Cedric: author's other books
Who wrote Models and Algorithms for Genome Evolution? Find out the surname, the name of the author of the book and a list of all author's works by series.







Models and Algorithms for Genome Evolution — read online for free the complete book (whole text) full work
Below is the text of the book, divided by pages. System saving the place of the last page read, allows you to conveniently read the book "Models and Algorithms for Genome Evolution" online for free, without having to search again every time where you left off. Put a bookmark, and you can go to the page where you finished reading at any time.
Font size:
Interval:
Bookmark:
Emergence of Standard Algorithms
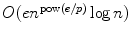 expected-time algorithm for finding all e -matches to a string of length p in a text of length n was completed in 1991. The function
expected-time algorithm for finding all e -matches to a string of length p in a text of length n was completed in 1991. The function 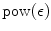 is 0 for =0 and concave increasing, so the algorithm is truly sublinear in that its running time is O ( n c ) for c <1 for sufficiently small. This paper reviews the history and the unfolding of the basic concepts, and it attempts to intuitively describe the deeper result whose time complexity, to this authors knowledge, has yet to be improved upon.
is 0 for =0 and concave increasing, so the algorithm is truly sublinear in that its running time is O ( n c ) for c <1 for sufficiently small. This paper reviews the history and the unfolding of the basic concepts, and it attempts to intuitively describe the deeper result whose time complexity, to this authors knowledge, has yet to be improved upon.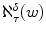 is the set of all strings v whose best alignment with w under scoring scheme is less than , i.e. { v :( v , w )}.
is the set of all strings v whose best alignment with w under scoring scheme is less than , i.e. { v :( v , w )}.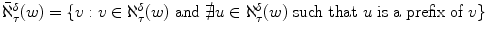 . For our example, the condensed 1-neighborhood of abba is
. For our example, the condensed 1-neighborhood of abba is 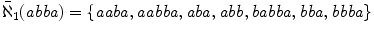 , a considerably smaller set of strings.
, a considerably smaller set of strings.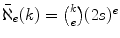 . Thus in our example we can filter the text by looking for one of 800 strings of length 20. Which filter is better? The probability of a random false positive for the k -mer filter is 10/ s 10 and for the neighborhood filter it is 800/ s 20. Thus the later filter produces s 10/80 fewer false positives. If s is 4 (e.g. the DNA alphabet) and n is 3109 (e.g. the size of the human genome) then the neighborhood filter produces 13,000 times fewer false positives, and reports in expectation 2.18 false positive, whereas the k -mer filter reports over 28,600!
. Thus in our example we can filter the text by looking for one of 800 strings of length 20. Which filter is better? The probability of a random false positive for the k -mer filter is 10/ s 10 and for the neighborhood filter it is 800/ s 20. Thus the later filter produces s 10/80 fewer false positives. If s is 4 (e.g. the DNA alphabet) and n is 3109 (e.g. the size of the human genome) then the neighborhood filter produces 13,000 times fewer false positives, and reports in expectation 2.18 false positive, whereas the k -mer filter reports over 28,600!Font size:
Interval:
Bookmark:
Similar books «Models and Algorithms for Genome Evolution»
Look at similar books to Models and Algorithms for Genome Evolution. We have selected literature similar in name and meaning in the hope of providing readers with more options to find new, interesting, not yet read works.
Discussion, reviews of the book Models and Algorithms for Genome Evolution and just readers' own opinions. Leave your comments, write what you think about the work, its meaning or the main characters. Specify what exactly you liked and what you didn't like, and why you think so.