Georg F Hoffmann Johannes Zschocke - Inherited Metabolic Diseases
Here you can read online Georg F Hoffmann Johannes Zschocke - Inherited Metabolic Diseases full text of the book (entire story) in english for free. Download pdf and epub, get meaning, cover and reviews about this ebook. City: Berlin, year: 2010, publisher: Springer Verlag Berlin Heidelberg, genre: Children. Description of the work, (preface) as well as reviews are available. Best literature library LitArk.com created for fans of good reading and offers a wide selection of genres:
Romance novel
Science fiction
Adventure
Detective
Science
History
Home and family
Prose
Art
Politics
Computer
Non-fiction
Religion
Business
Children
Humor
Choose a favorite category and find really read worthwhile books. Enjoy immersion in the world of imagination, feel the emotions of the characters or learn something new for yourself, make an fascinating discovery.

- Book:Inherited Metabolic Diseases
- Author:
- Publisher:Springer Verlag Berlin Heidelberg
- Genre:
- Year:2010
- City:Berlin
- Rating:4 / 5
- Favourites:Add to favourites
- Your mark:
Inherited Metabolic Diseases: summary, description and annotation
We offer to read an annotation, description, summary or preface (depends on what the author of the book "Inherited Metabolic Diseases" wrote himself). If you haven't found the necessary information about the book — write in the comments, we will try to find it.
Further, it offers helpful advice for emergency situations, such as hypoglycemia, hyperammonemia, lactic acidosis or acute encephalopathy. Five different indices allow a quick but complete orientation for common important constellations. Last but not least, it has an appendix with a guide to rapid differential diagnosis of signs and symptoms and when not to suspect metabolic disease and is accompanied by a CD-ROM including the whole content of the book as well as interactive tables and links to the respective OMIM page as well as enzyme and protein information. It will help physicians to diagnose patients they may otherwise fail to diagnose and to reduce unnecessary referrals. For metabolic and genetic specialists especially the indices will be helpful as a quick look when being called for advice. It has all it needs to become a gold standard defining the clinical practice in this field.
Georg F Hoffmann Johannes Zschocke: author's other books
Who wrote Inherited Metabolic Diseases? Find out the surname, the name of the author of the book and a list of all author's works by series.












Inherited Metabolic Diseases — read online for free the complete book (whole text) full work
Below is the text of the book, divided by pages. System saving the place of the last page read, allows you to conveniently read the book "Inherited Metabolic Diseases" online for free, without having to search again every time where you left off. Put a bookmark, and you can go to the page where you finished reading at any time.
Font size:
Interval:
Bookmark:
Introduction to Inborn Errors of Metabolism
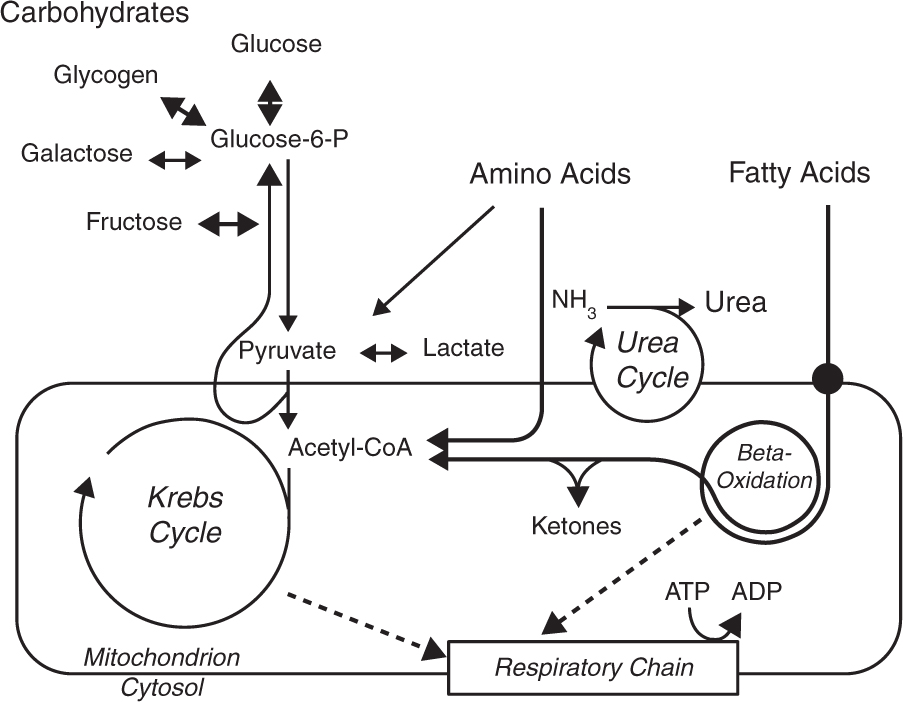
- The classical inborn errors of metabolism are defects in enzymes of the metabolism of amino acids, carbohydrates and fatty acids or in mito-chondrial energy metabolism (Fig. A1.1).
- Disorders of intermediary metabolism are often dynamic, they fluctuate with changes in the metabolic state of the patient and frequently allow successful therapeutic intervention.
- Most disorders of intermediary metabolism are readily diagnosed through basic metabolic investigations which include blood gases, glucose, lactate, ammonia, plasma amino acids, urinary organic acids and an acylcarnitine profile.
Font size:
Interval:
Bookmark:
Similar books «Inherited Metabolic Diseases»
Look at similar books to Inherited Metabolic Diseases. We have selected literature similar in name and meaning in the hope of providing readers with more options to find new, interesting, not yet read works.
Discussion, reviews of the book Inherited Metabolic Diseases and just readers' own opinions. Leave your comments, write what you think about the work, its meaning or the main characters. Specify what exactly you liked and what you didn't like, and why you think so.