Kim Jelfs - Computer Simulation of Porous Materials: Current Approaches and Future Opportunities
Here you can read online Kim Jelfs - Computer Simulation of Porous Materials: Current Approaches and Future Opportunities full text of the book (entire story) in english for free. Download pdf and epub, get meaning, cover and reviews about this ebook. year: 2021, publisher: Royal Society of Chemistry, genre: Home and family. Description of the work, (preface) as well as reviews are available. Best literature library LitArk.com created for fans of good reading and offers a wide selection of genres:
Romance novel
Science fiction
Adventure
Detective
Science
History
Home and family
Prose
Art
Politics
Computer
Non-fiction
Religion
Business
Children
Humor
Choose a favorite category and find really read worthwhile books. Enjoy immersion in the world of imagination, feel the emotions of the characters or learn something new for yourself, make an fascinating discovery.

- Book:Computer Simulation of Porous Materials: Current Approaches and Future Opportunities
- Author:
- Publisher:Royal Society of Chemistry
- Genre:
- Year:2021
- Rating:4 / 5
- Favourites:Add to favourites
- Your mark:
Computer Simulation of Porous Materials: Current Approaches and Future Opportunities: summary, description and annotation
We offer to read an annotation, description, summary or preface (depends on what the author of the book "Computer Simulation of Porous Materials: Current Approaches and Future Opportunities" wrote himself). If you haven't found the necessary information about the book — write in the comments, we will try to find it.
Kim Jelfs: author's other books
Who wrote Computer Simulation of Porous Materials: Current Approaches and Future Opportunities? Find out the surname, the name of the author of the book and a list of all author's works by series.










Computer Simulation of Porous Materials: Current Approaches and Future Opportunities — read online for free the complete book (whole text) full work
Below is the text of the book, divided by pages. System saving the place of the last page read, allows you to conveniently read the book "Computer Simulation of Porous Materials: Current Approaches and Future Opportunities" online for free, without having to search again every time where you left off. Put a bookmark, and you can go to the page where you finished reading at any time.
Font size:
Interval:
Bookmark:
Chapter 1
Introduction to Computational Modelling of Microporous Materials
Kim E. Jelfs
a Department of Chemistry, Molecular Sciences Research Hub, Imperial College London White City Campus, Wood Lane W12 0BZ United Kingdom Email:
Porous materials are materials that have void spaces within them that can be occupied by guests to produce a range of different properties and functionalities. Depending upon the size of their pores, porous materials are classified as microporous (pores smaller than 2 nm), mesoporous (pores between 2 and 50 nm) or macroporous (pores larger than 50 nm). Here, we will focus mostly upon microporous materials, as these pores can host small molecules and atoms and therefore provide functionality across a range of applications from encapsulation, separation, catalysis, sensing and more. As a note on terminology, the term sorption is generally used in the discussion of porous materials as these materials will both adsorb guests onto surfaces as well as absorb guests into the material's bulk.
The computational modelling of materials is increasingly widespread, and porous materials modelling is no exception to this. Computational modelling in porous materials has already had decades of application, especially in the field of zeolites, with significant impact in rationalising the structure and property of the materials, frequently providing additional atomic-level insight into the materials that could not be uncovered through experimental characterisation alone. This a posteriori rationalisation of experimental observations built a foundation upon which many predictive studies have been built, for example, screening materials in advance for useful properties prior to experimental realisation. A commonly used term is computational materials design, but we urge caution in not overusing the term design when a more appropriate term is screening or discovery. We should leave the term design for those occasions when our insight, knowledge and software truly have enabled design. Computational screening, with consideration of both whether a hypothetical material can be experimentally realised and have target properties, is powerful in its own right.
Ever increasing computing power, increasing computational literacy and programming ability, combined with open-source data and algorithms hold great potential for the further impact of modelling in the field of porous materials. As in all computational fields, there is significant emphasis on the potential for advances through the use of artificial intelligence, notably machine learning. There is enormous value in all porous material scientists understanding, even if not directly applying, the potential of computational modelling for the advancement of their own research. Beyond technological advancements, we can also expect an increasing focus on understanding and predicting more complex behaviour, for example, consideration of multifunctionality in a material, such as the interplay of porosity with optoelectronic properties, and the consideration of the importance and utilisation of defects as an opportunity for property tuning.
In this book, the key foundations for porous materials modelling are laid out, along with the major areas where modelling is applied, successes so far, as well as the future prospects for the field. In this chapter (Chapter 1), the key classes of microporous materials are introduced, along with the applications of porous materials. This is followed by the introduction of the key types of modelling approaches that are applied in the area of porous materials. This chapter should therefore provide the foundation for accessing the following chapters that focus on specific application areas of porous materials modelling. Chapter 2 (Addicoat) outlines the approaches and successes in the structure prediction of porous materials, covering both bottom-up approaches from building blocks and top-down approaches from libraries of topological nets. Examples are discussed from zeolites, metalorganic frameworks, polymers and porous molecules. With a known structure of a material, either from a simulation or an experimental crystal structure, many further simulations analysing properties and function can be performed. Chapter 3 (Evans) discusses the analysis of the mechanical properties of porous materials. How to analyse the strength and flexibility of porous materials is covered, along with the potential to uncover unusual properties, such as negative thermal expansion. The modelling of both sorption and diffusion behaviour in porous materials is covered next (Glover and Besley, Chapter 4). This spans sorption simulations of guest uptake, pore analysis, and molecular dynamics and enhanced sampling techniques for guest diffusion. Modelling the spectroscopic and catalytic properties of porous materials is then covered (Morales-Vidal and Ortuo, Chapter 5), including a discussion of how to choose appropriate methods, followed by examples of mechanistic investigations, focused upon metalorganic frameworks and covalent organic frameworks. Finally, the future for the field, in particular through the application of artificial intelligence techniques, is covered (Jensen and Olivetti, Chapter 6).
There are a large range of microporous materials and these can have a variety of chemical compositions, whether inorganic or organic in nature, as well as hybrid materials containing both inorganic and organic components. Microporous materials also vary by their degree of crystallinity, with some having both short- and long-range order, and amorphous porous materials lacking any long-range order at all. In this section, the key classes of porous material that are discussed in later chapters will be introduced.
Zeolites are naturally occurring crystalline aluminosilicate minerals that are typically found in regions where there has been volcanic activity. The name zeolite originates from how the rock appeared to boil upon heating, with the ancient Greek zein for to boil and lithus for rock, so literally zeolite means boiling rock. It was not until the 1940s that Barrer found a method to synthesise zeolites in a laboratory. Barrer was able to synthesise zeolites via mimicking the conditions where they form naturally, in particular the high temperature and pressure, along with organic template molecules that would direct the formation to a targeted zeolite structure. There are now more than two hundred zeolite structures that are either naturally occurring or have been synthesised in the laboratory.
Zeolites are composed of [SiO4]4 and [AlO4]5 tetrahedra, and these primary building units, known as T-sites, are linked by the bridging oxygen atoms to produce corner-sharing tetrahedra. Through the multitude of ways that these tetrahedra can be linked and arranged to form crystalline arrays, there are millions of hypothetical zeolite topologies. There are a series of what are known as secondary building units or SBUs that can be used to characterise a zeolite topology. Examples of SBUs include n-membered rings (where n = 3, 4, 5, 6), and then the equivalent doubled rings, double 4-ring (D4R), double 5-ring (D5R), double 6-ring (D6R) and so on that have n bridging oxygens linking the two rings, as well as a small number of other SBUs linking different sized rings. Each zeolite has a three letter framework type code assigned by the International Zeolite Association,.
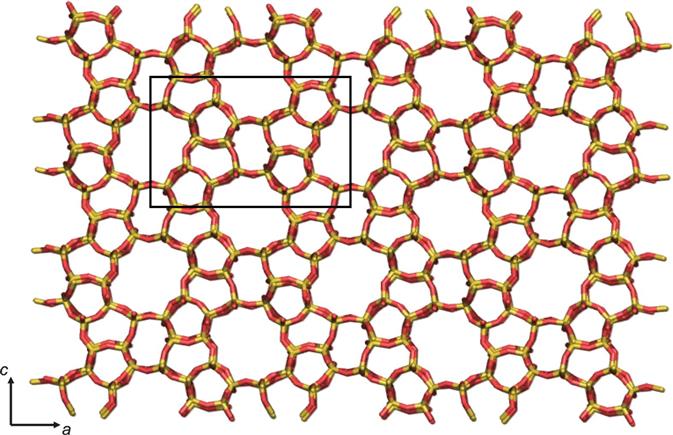
Figure 1.1 An example of a zeolite structure. This is the zeolite MFI viewed down the
Next pageFont size:
Interval:
Bookmark:
Similar books «Computer Simulation of Porous Materials: Current Approaches and Future Opportunities»
Look at similar books to Computer Simulation of Porous Materials: Current Approaches and Future Opportunities. We have selected literature similar in name and meaning in the hope of providing readers with more options to find new, interesting, not yet read works.
Discussion, reviews of the book Computer Simulation of Porous Materials: Current Approaches and Future Opportunities and just readers' own opinions. Leave your comments, write what you think about the work, its meaning or the main characters. Specify what exactly you liked and what you didn't like, and why you think so.