1. Genodermatoses
Fabry Disease
Introduction
Fabry disease is an X-linked recessive disorder caused by a deficiency in the activity of alpha-galactosidase A, an enzyme responsible for breaking down sphingoglycolipids [].
Epidemiology
The disease affects all ethnicities, and the incidence has been estimated between 1 in 40,000 and 1 in 117,000 live male births [].
Genetics
Fabry disease results from mutations in the gene GLA encoding the lysosomal enzyme alpha-galactosidase A, and this gene is found on the long arm of chromosome X [].
Attempts to correlate genotype and phenotype have not been consistent [].
Most mutations in GLA are inherited, rather than spontaneous; therefore, most patients will have affected male or carrier female family members [].
Patients may have variable levels of enzyme activity, which can impact the degree of clinical severity. However, a fairly low level of activity (above 10 %) is generally sufficient to prevent clinical disease [].
Cutaneous Findings
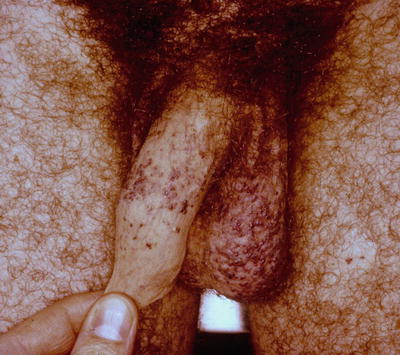
Angiokeratoma corporis diffusum is the hallmark skin manifestation of Fabry disease and comprises nonblanching red to black discrete vascular papules that range in size from 1 to 10 mm and occur preferentially in a bathing trunk distribution, becoming more generalized and even involving mucosa over time [].
Fig. 1.1
Angiokeratomas associated with Fabry disease. Note the multiple, discrete, nonblanching, vascular papules on the penile shaft and scrotum. The differential diagnosis would also include angiokeratomas of Fordyce, which are common growths in the genital area in elderly patients and have no systemic associations. Photo courtesy of Dirk Elston, M.D.
Angiokeratoma corporis diffusum can occur in the setting of other lysosomal storage diseases (fucosidosis, sialidosis, etc.) and sometimes in the absence of other enzymatic abnormalities, thus is not pathognomonic for Fabry disease []. Thus skin biopsy can be important in cementing the diagnosis.
Routine histopathologic examination of an angiokeratoma fixed in formalin and stained with hematoxylin and eosin reveals a dilated, thin-walled vascular proliferation (sometimes filled with red blood cells) of the papillary dermis that is enveloped by an acanthotic epidermis with overlying hyperkeratosis of the stratum corneum [].
While angiokeratomas are the most often encountered cutaneous vascular lesion in Fabry patients, telangiectasias and cherry angiomas have been reported to occur as well [].
Other skin manifestations can include abnormalities of sweating, lymphedema, and characteristic facies. Hypohidrosis is reported by patients more commonly than anhidrosis or hyperhidrosis, but all have been documented to occur [].
Renal Manifestations
Fabry nephropathy is caused by glomerular and renal vascular deposition of neutral glycosphingolipids, which have been demonstrated as early as in utero [].
In patients with rapidly declining renal function, renal ultrasound should be performed to rule out other possible causes, such as renal cysts [].
Renal biopsy can be undertaken in those with known Fabry disease and in whom it is unclear whether enzyme replacement therapy should be started. If electron microscopic examination reveals GL-3 within podocytes and vascular endothelial cells, enzyme replacement therapy may be indicated [].
A less invasive test to assess for Fabry disease of the kidney is urine microscopy. When viewing urine sediment via polarized microscope, autofluorescent, birefringent Maltese cross lipid particles can be seen. Different morphologies of Maltese cross particles have been defined, with those having a lamellated appearance with surface protrusions being most sensitive and specific for Fabry disease [].
Cardiovascular Manifestations
Glycosphingolipids also accumulate in various cardiac tissues, leading to dysfunction. Myocytes, valves, conduction tissue, and vascular endothelium may all be affected, and the resulting presenting symptoms include dyspnea, syncope, palpitations, or angina [].
The cardiomyopathy of Fabry disease is classified as restrictive, due to infiltration of glycosphingolipids within the cardiac myocytes [].
Dysrhythmias occur as a result of glycosphingolipid deposition within the conduction system or by atrial dilatation and ischemia due to left ventricular hypertrophy [].
Over half of Fabry patients complain of anginal chest pain, with glycosphingolipid deposition within cardiac endothelial cells playing a role in vascular dysfunction and ultimately in myocardial injury [].
Valvular insufficiency most often manifests as mitral valve regurgitation, but patients do not generally require valve replacement [].
Neurologic Manifestations
The peripheral, central, and autonomic nervous systems may all be affected in Fabry disease.
Fabry pain crises, occurring as attacks of neuropathic pain, may be the first manifestation of the condition. They can begin in early childhood with episodes of severe, burning pain in the distal extremities, brought on by stress, infections, or changes in temperature and lasting minutes to days [].
Cerebrovascular disease, caused by glycosphingolipid deposition within cerebral vessels, is a common cause of morbidity and mortality in patients with Fabry disease [].
Involvement of the autonomic nervous system manifests as abnormalities of sweating and gastrointestinal complaints [].
Ophthalmologic Manifestations
Cornea verticillata, bilateral, whorl-shaped corneal opacities diagnosed by slit-lamp examination, is the characteristic eye finding in patients with Fabry disease and can be seen in nearly all patients by age 10 [].
Other Organ Involvement
Other symptoms reported in Fabry patients are diverse and can include:
Gastrointestinal disturbances: Nausea, vomiting, diarrhea, abdominal pain []
Tinnitus, hearing loss, vertigo []
Obstructive and restrictive pulmonary disease: Dyspnea, cough, wheezing []
Psychiatric disorders: Depression, substance abuse, suicide []
Sexual dysfunction: Delayed puberty, impotence, priapism, infertility []
Diagnosis
Signs and symptoms that suggest the diagnosis of Fabry disease are as follows: attacks of pain in distal extremities triggered by physiological or psychological stress, cutaneous angiokeratomas, proteinuria or renal failure of unknown cause, left ventricular hypertrophy, stroke, characteristic corneal opacities, and unexplained gastrointestinal complaints [].
In affected males, the diagnosis can be confirmed by measuring alpha-galactosidase A activity in plasma or peripheral leukocytes [].